
【药化经典】PI3K抑制剂:综述和新的开发策略
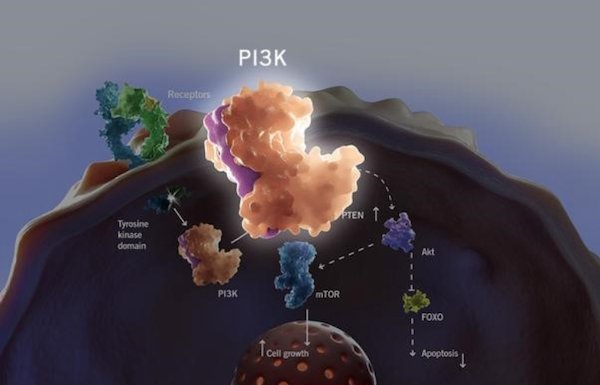
磷脂酰肌醇3-激酶( phosphoinositide 3-kinase,PI3Ks) 途径是人类中最常被激活的信号通路之一,几乎介导50%的恶性肿瘤的发生。PI3K是一种胞内脂质磷酸激酶,催化肌醇第3位的磷酸化。PI3K是由一个催化亚基p110和一个调节亚基p85构成的异二聚体,具有SH2结构域 (Src Homology 2 domain)。通过SH2结构域,PI3K可与其它蛋白的磷酸化酪氨酸残基结合,从而被募集到质膜,使其催化亚基靠近质膜内小叶的磷脂酰肌醇。在膜脂代谢过程中,PI3K催化PI-4-P(PIP)生成PI-3,4-P2(PIP2),催化PI-4,5-P2(PIP2)生成PI-3,4,5-P3(PIP3)。这些与膜结合的PI-3-P为多种信号转导蛋白提供了锚定位点,进而介导多种下游信号通路。例如活化Akt/PKB、mTOR激酶等下游通路。
根据PI3K的结构特点和底物分子可将其分为Ⅰ型、Ⅱ型、Ⅲ型3 类。Ⅰ型分为IA型和IB型, IA型由受体酪氨酸激活,根据催化亚基p110的不同又可分为PI3Kα、PI3Kβ 和PI3Kδ 3个亚型;IB型即为PI3Kγ 亚型。IA型PI3K与肿瘤的发生发展密切相关,其中编译PI3Kα的基因PIK3CA是肿瘤中最常见的突变,PIK3CA突变后在异常激活PI3Kα的同时,还能抑制抑癌基因PTEN的表达,因此PI3Kα成为抗癌药物研发中极为重要的靶点
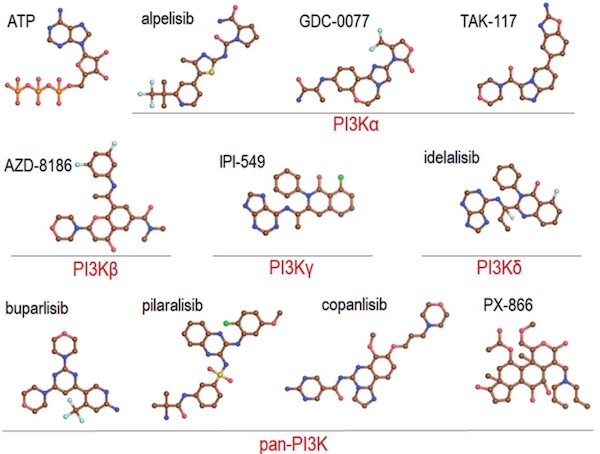
目前,已有许多PI3K抑制剂批准上市或进入临床研究。泛PI3K抑制剂,如buparlisib、pictilisb (GDC-0941)、copanlisib等,泛PI3K抑制剂可以作用于所有I型的PI3K亚型,因此副作用与毒性较大,使得它们的临床进展缓慢。选择性作用于某个PI3K亚型的抑制剂具有更小的毒副作用,已有部分选择性PI3K抑制剂获批上市。Alpelisib是FDA批准的第一个PI3Kα抑制剂用于乳腺癌治疗,当Alpelisib靶向PI3Kα时,观察到此药可以有效地降低特定突变组合的信号强度。虽然该药被归类为PI3Kα特异性药物,但通常会观察到严重的浓度依赖性副作用和耐药性。主要原因是该药物和以前开发的药物一样,都是ATP竞争性的,而PI3K各亚型之间的ATP结合位点几乎相同,当给药浓度增加,不可避免地对其它亚型产生抑制作用。因此降低副作用,对PI3K抑制剂的开发与癌症治疗具有重要意义。
本文作者Ruth Nussinov回顾了PI3K抑制剂,基于其课题组前期研究概述了PI3Kα激活的机制、致癌突变及其联合突变。最终提出了两种新的可能的PI3Kα抑制剂开发策略。(1) PI3Kα正构和别构抑制剂的联合使用;(2) 开发挽救突变位点的别构抑制剂。
PI3Kα的激活机制
PI3K激活发生在质膜上,它是通过与RTK、RAS和膜上C-末端的磷酸化基序相互作用而介导的。如下图展示了突变位点导致PI3K激活的过程。
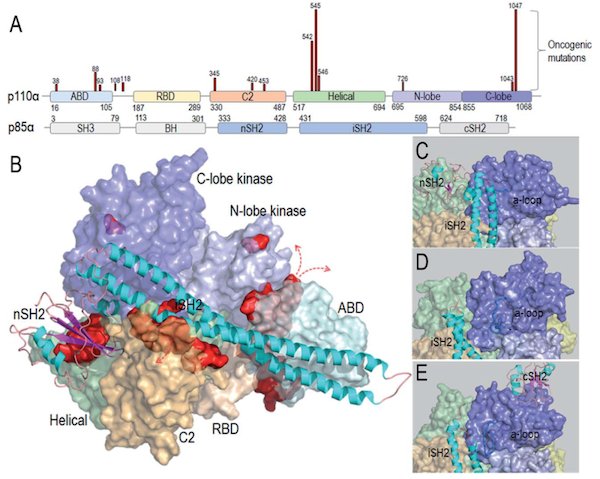
上图A:热点突变E542K和E545K模拟RTK在PI3Kα的解除自抑制中的作用机制,这一作用暴露了PI3Kα活性位点。H1047R模拟了RAS在PI3Kα激活过程的作用机制。这两处热点突变常伴随E453Q/K、E726K和M1043V/I等弱突变共同激活PI3Kα。大多数弱突变都远离p85α亚基的自抑制结构域nSH2和催化位点,推断它们可能在激活PI3Kα时起别构位点作用。
图B:热点突变(深色部分)主要发生在螺旋域和激酶结构域,弱突变发生在ABD和C2结构域的表面。ABD和C2中的iSH2结构域在激活的PI3Kα结构中活跃表达。
图C:含有自抑制区域的nSH2的PI3Kα的结构处于非活性构象
图D:PI3Kα在移除nSH2后并伴有a-loop时处于激活状态。
图E:移除nSH2结构域的PI3Kβ只具有部分活性。说明PI3Kβ被nSH2和cSH2同时抑制。这意味着PI3Kα和PI3Kβ亚型自抑制区域不同。
受到KRas4B共价抑制剂开发过程的启发,可以尝试发现新的PI3Kα的别构口袋,来开发PI3Kα的别构抑制剂,在维持抑制活性的同时提高化合物对PI3K亚型的选择性。
PI3Kα与Alpelisib的相互作用解析

PI3K的ATP结合位点位于激酶结构域的两个片段之间,由铰链隔开。I型PI3K的ATP结合位点高度同源,仅在边缘处有几个残基不同。这些残基可分为分为两个区域,第一个是位于铰链附近的四个残基组成的可变区,第二个是位于P-loop处的较小的可变区。对于PI3Kα,铰链区可以提高ATP抑制剂的选择性。如图,Alpelisib结合激酶结构域中的ATP口袋,并与P-loop和铰链区相互作用。其中与Alpelisib接触的残基大多是保守的,只有铰链区五个可变残基(852RNSH855和Q859)。在这里,作者重点研究两个残基R852和Q859,因为其他三个残基(NSH)的侧链暴露于溶剂中,几乎不与Alpelisib的相互作用。PI3Kα的Q859与Alpelisib形成双氢键(图C),这表明并证实了其在亚型选择性中的重要性。此残基在PI3Kβ(D856)和PI3Kδ(N836)中变得更短,而PI3Kγ中的K890仍然足够长,足以与Alpelisib进行氢键相互作用。PI3Kα中的R852与激酶结构域N-lobe中的E798形成盐桥,在PI3Kγ(K831-E814)中观察到类似的盐桥。PI3Kγ的L829还与M762和W760建立疏水相互作用。而PI3Kβ中S849由于较短,则完全没有类似的相互作用。
- 上一篇:方达控股收购ACME 扩展合成与药物化学及工艺研究
- 下一篇:没有了